Fibrose kystique

La mucoviscidose est une maladie qui provoque une accumulation de mucus épais et collant dans les poumons, le tube digestif et d'autres parties du corps. C'est l'une des maladies pulmonaires chroniques les plus courantes chez les enfants et les jeunes adultes. C'est une maladie mortelle.
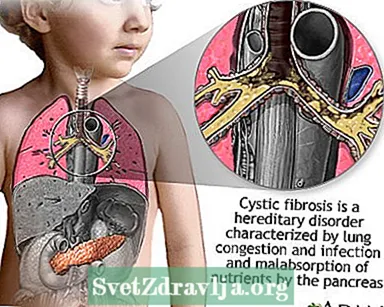
La mucoviscidose (FK) est une maladie qui se transmet dans les familles. Elle est causée par un gène défectueux qui fait que le corps produit un liquide anormalement épais et collant, appelé mucus. Ce mucus s'accumule dans les voies respiratoires des poumons et dans le pancréas.
L'accumulation de mucus entraîne des infections pulmonaires potentiellement mortelles et de graves problèmes de digestion. La maladie peut également affecter les glandes sudoripares et le système reproducteur de l'homme.
De nombreuses personnes sont porteuses d'un gène CF, mais ne présentent aucun symptôme. En effet, une personne fibro-kystique doit hériter de 2 gènes défectueux, 1 de chaque parent. Certains Américains ont le gène CF. Il est plus fréquent chez les descendants d'Europe du Nord ou d'Europe centrale.
La plupart des enfants atteints de mucoviscidose sont diagnostiqués avant l'âge de 2 ans, d'autant plus que le dépistage néonatal est effectué aux États-Unis. Pour un petit nombre, la maladie n'est détectée qu'à l'âge de 18 ans ou plus. Ces enfants ont souvent une forme bénigne de la maladie.
Les symptômes chez les nouveau-nés peuvent inclure :
- Croissance retardée
- Ne pas prendre de poids normalement pendant l'enfance
- Pas de selles dans les 24 à 48 premières heures de vie
- Peau au goût salé
Les symptômes liés à la fonction intestinale peuvent inclure :
- Douleur au ventre due à une constipation sévère
- Augmentation des gaz, ballonnements ou ventre qui semble gonflé (distendu)
- Nausées et perte d'appétit
- Selles pâles ou de couleur argile, nauséabondes, contenant du mucus ou flottant
- Perte de poids
Les symptômes liés aux poumons et aux sinus peuvent inclure :
- Toux ou augmentation du mucus dans les sinus ou les poumons
- Fatigue
- Congestion nasale causée par des polypes nasaux
- Épisodes répétés de pneumonie (les symptômes de pneumonie chez une personne atteinte de mucoviscidose incluent fièvre, toux accrue et essoufflement, augmentation du mucus et perte d'appétit)
- Douleur ou pression des sinus causée par une infection ou des polypes
Symptômes qui peuvent être remarqués plus tard dans la vie :
- Infertilité (chez les hommes)
- Inflammation répétée du pancréas (pancréatite)
- Symptômes respiratoires
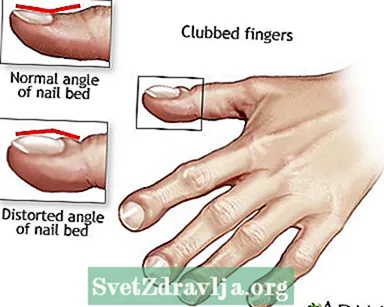
- Doigts matraqués
Un test sanguin est effectué pour aider à détecter la mucoviscidose. Le test recherche des changements dans le gène CF. Les autres tests utilisés pour diagnostiquer la mucoviscidose comprennent :
- Le test immunoréactif au trypsinogène (IRT) est un test standard de dépistage néonatal de la mucoviscidose. Un niveau élevé d'IRT suggère une possible FC et nécessite des tests supplémentaires.
- Le test de chlorure de sueur est le test de diagnostic standard pour la mucoviscidose. Un niveau élevé de sel dans la sueur de la personne est un signe de la maladie.
D'autres tests qui identifient les problèmes qui peuvent être liés à la mucoviscidose comprennent :
- Radiographie pulmonaire ou tomodensitométrie
- Test de graisse fécale
- Tests de fonction pulmonaire
- Mesure de la fonction pancréatique (élastase pancréatique des selles)
- Test de stimulation de la sécrétine
- Trypsine et chymotrypsine dans les selles
- Série GI supérieure et intestin grêle
- Cultures pulmonaires (obtenues par crachats, bronchoscopie ou écouvillonnage de gorge)
Un diagnostic précoce de la mucoviscidose et un plan de traitement peuvent améliorer à la fois la survie et la qualité de vie. Le suivi et le contrôle sont très importants. Dans la mesure du possible, les soins devraient être reçus dans une clinique spécialisée en fibrose kystique. Lorsque les enfants atteignent l'âge adulte, ils doivent être transférés dans un centre spécialisé en fibrose kystique pour adultes.
Le traitement des problèmes pulmonaires comprend :
- Antibiotiques pour prévenir et traiter les infections des poumons et des sinus. Ils peuvent être pris par voie orale, administrés dans les veines ou par des traitements respiratoires. Les personnes fibro-kystiques peuvent prendre des antibiotiques uniquement au besoin ou tout le temps. Les doses sont souvent supérieures à la normale.
- Médicaments inhalés pour aider à ouvrir les voies respiratoires.
- D'autres médicaments qui sont administrés par un traitement respiratoire pour fluidifier le mucus et le rendre plus facile à tousser sont la thérapie enzymatique DNAse et les solutions salines hautement concentrées (solution saline hypertonique).
- Vaccin contre la grippe et vaccin antipneumococcique polyosidique (VPP) chaque année (demandez à votre fournisseur de soins de santé).
- La transplantation pulmonaire est une option dans certains cas.
- Une oxygénothérapie peut être nécessaire à mesure que la maladie pulmonaire s'aggrave.
Les problèmes pulmonaires sont également traités avec des thérapies pour fluidifier le mucus. Cela permet de tousser plus facilement le mucus des poumons.
Ces méthodes incluent :
- Activité ou exercice qui vous fait respirer profondément
- Appareils utilisés pendant la journée pour aider à dégager les voies respiratoires de trop de mucus
- Percussion thoracique manuelle (ou physiothérapie thoracique), dans laquelle un membre de la famille ou un thérapeute frappe légèrement la poitrine, le dos et la zone sous les bras de la personne
Le traitement des problèmes intestinaux et nutritionnels peut inclure :
- Un régime spécial riche en protéines et en calories pour les enfants plus âgés et les adultes
- Enzymes pancréatiques pour aider à absorber les graisses et les protéines, qui sont prises à chaque repas
- Suppléments vitaminiques, en particulier les vitamines A, D, E et K
- Votre fournisseur peut vous conseiller d'autres traitements si vous avez des selles très dures
L'ivacaftor, le lumacaftor, le tézacaftor et l'élexacaftor sont des médicaments qui traitent certains types de mucoviscidose.
- Ils améliorent la fonction de l'un des gènes défectueux qui cause la mucoviscidose.
- Jusqu'à 90 % des patients atteints de mucoviscidose et éligibles pour un ou plusieurs de ces médicaments seuls ou en association.
- En conséquence, il y a moins d'accumulation de mucus épais dans les poumons. D'autres symptômes de la mucoviscidose sont également améliorés.
Les soins et le suivi à domicile doivent inclure :
- Éviter la fumée, la poussière, la saleté, les vapeurs, les produits chimiques ménagers, la fumée de cheminée et la moisissure.
- Donner beaucoup de liquides, en particulier aux nourrissons et aux enfants par temps chaud, en cas de diarrhée ou de selles molles, ou pendant une activité physique supplémentaire.
- Faire de l'exercice 2 ou 3 fois par semaine. La natation, le jogging et le vélo sont de bonnes options.
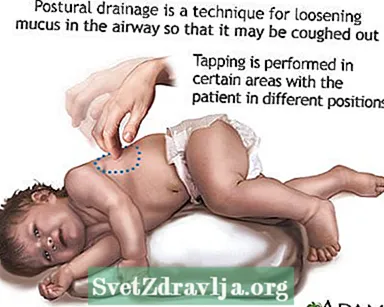
- Éliminer ou faire remonter le mucus ou les sécrétions des voies respiratoires. Cela doit être fait 1 à 4 fois par jour. Les patients, les familles et les soignants doivent apprendre à pratiquer la percussion thoracique et le drainage postural pour aider à garder les voies respiratoires dégagées.
- Aucun contact avec d'autres personnes fibro-kystiques n'est recommandé car ils peuvent échanger des infections (ne s'applique pas aux membres de la famille).
Vous pouvez soulager le stress de la maladie en vous joignant à un groupe de soutien pour la fibrose kystique. Partager avec d'autres personnes qui ont des expériences et des problèmes communs peut aider votre famille à ne pas se sentir seule.
La plupart des enfants atteints de mucoviscidose restent en bonne santé jusqu'à ce qu'ils atteignent l'âge adulte. Ils peuvent participer à la plupart des activités et aller à l'école. De nombreux jeunes adultes fibro-kystiques terminent leurs études collégiales ou trouvent un emploi.
La maladie pulmonaire finit par s'aggraver au point où la personne est handicapée. Aujourd'hui, la durée de vie moyenne des personnes fibro-kystiques qui atteignent l'âge adulte est d'environ 44 ans.
La mort est le plus souvent causée par des complications pulmonaires.
La complication la plus fréquente est l'infection respiratoire chronique.
Les autres complications comprennent :
- Problèmes intestinaux, tels que calculs biliaires, blocage intestinal et prolapsus rectal
- Tousser du sang
- Insuffisance respiratoire chronique
- Diabète
- Infertilité
- Maladie du foie ou insuffisance hépatique, pancréatite, cirrhose biliaire
- Malnutrition
- Polypes nasaux et sinusite
- Ostéoporose et arthrite
- Pneumonie qui revient sans cesse
- Pneumothorax
- Insuffisance cardiaque droite (cœur pulmonaire)
- Cancer colorectal
Appelez votre prestataire si un nourrisson ou un enfant présente des symptômes de mucoviscidose et éprouve :
- Fièvre, toux accrue, modifications des expectorations ou du sang dans les expectorations, perte d'appétit ou autres signes de pneumonie
- Perte de poids accrue
- Selles plus fréquentes ou selles nauséabondes ou contenant plus de mucus
- Ventre gonflé ou ballonnements accrus
Appelez votre fournisseur si une personne fibro-kystique développe de nouveaux symptômes ou si les symptômes s'aggravent, notamment des difficultés respiratoires graves ou des crachats de sang.
La mucoviscidose ne peut pas être empêchée. Le dépistage des personnes ayant des antécédents familiaux de la maladie peut détecter le gène CF chez de nombreux porteurs.
FC
- Nutrition entérale - enfant - gestion des problèmes
- Sonde d'alimentation pour gastrostomie - bolus
- Comment respirer quand on est essoufflé
- Tube d'alimentation de jéjunostomie
- Drainage postural
 Sortir en boite
Sortir en boite Drainage postural
Drainage postural Doigts matraqués
Doigts matraqués Fibrose kystique
Fibrose kystique
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor chez les sujets atteints de mucoviscidose et F508del/F508del-CFTR ou F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-224. PMID : 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Fibrose kystique. Dans : Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, éd. Manuel Nelson de pédiatrie. 21e éd. Philadelphie, Pennsylvanie : Elsevier ; 2020 : chapitre 432.
Farrell PM, White TB, Ren CL, et al. Diagnostic de la mucoviscidose : directives consensuelles de la Cystic Fibrosis Foundation. J Pédiatre. 2017;181S:S4-S15.e1. PMID : 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effets du traitement par lumacaftor/ivacaftor sur la fonction CFTR chez les patients homozygotes Phe508del atteints de mucoviscidose. Am J Respir Crit Care Med. 2018;197(11):1433-1442. PMID : 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Mucoviscidose. Dans : Goldman L, Schafer AI, éd. Médecine Goldman-Cecil. 26e éd. Philadelphie, Pennsylvanie : Elsevier ; 2020 : chap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibrose kystique. Dans : Broaddus VC, Mason RJ, Ernst JD, et al, eds. Manuel de médecine respiratoire de Murray et Nadel. 6e éd. Philadelphie, Pennsylvanie : Elsevier Saunders ; 2016 : chap. 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor chez les patients atteints de mucoviscidose homozygote pour phe508del. N Anglais J Med. 2017 ; 377(21): 2013-2023. PMID : 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Articles De Portail

Comment faire un gel doseur maison
